mag 16, 2023
Philips fornisce un aggiornamento sui risultati dei test completati per i dispositivi per la terapia del sonno CPAP/BiPAP
Amsterdam, Paesi Bassi – Royal Philips (NYSE: PHG; AEX: PHIA) ha presentato un aggiornamento sul programma completo di test e ricerca condotto dalla sua controllata Philips Respironics, al fine di valutare i potenziali rischi per la salute relativi alla schiuma fonoassorbente in poliuretano a base di poliestere (PE-PUR) presente in specifici dispositivi per la terapia del sonno e ventilatori in seguito all’avviso di sicurezza volontario del giugno 2021.*
Le valutazioni del rischio sono state ultimate per i dispositivi CPAP/BiPAP per la terapia del sonno oggetto dell’avviso di sicurezza*, ovvero i dispositivi DreamStation, System One e DreamStation Go di prima generazione, che rappresentano circa il 95% dei dispositivi registrati a livello globale. Le valutazioni si basano sui precedenti aggiornamenti rilasciati a dicembre 2021, giugno 2022 e dicembre 2022. Inoltre, sono stati completati test e analisi sui dispositivi DreamStation di prima generazione sottoposti a pulizia con ozono.
Metodi di test
Il programma di test e ricerca è stato condotto congiuntamente da cinque laboratori indipendenti e certificati, e i risultati sono stati esaminati e valutati da parte di esperti terzi qualificati e da Philips Respironics, oltre che da un comitato medico esterno.
I metodi di test applicati – compreso la pianificazione e l’esecuzione dei test e l’interpretazione dei risultati per le valutazioni del rischio ultimate - sono conformi agli standard industriali ISO 18562 [1,2] e ISO 10993 [3]. La progettazione dei metodi di test applicati è stata ulteriormente corroborata dal punto di vista scientifico, sulla base di un’attenta valutazione e mitigazione delle limitazioni intrinseche dei test. Ad esempio, sono stati eseguiti test su più dispositivi usati caratterizzati da una diversa durata di utilizzo da parte del paziente e da una degradazione visiva osservata della schiuma, nonché su schiuma invecchiata in laboratorio che era stata intenzionalmente degradata a diversi livelli. Nelle valutazioni del rischio sono state incluse ipotesi fortemente conservative. In calce al presente comunicato stampa vengono riportati ulteriori esempi.
“La nostra priorità è la salute e il benessere dei pazienti”, ha dichiarato Roy Jakobs, CEO di Royal Philips. “Ci siamo concentrati sul programma completo di test e ricerca al fine di ottenere maggiore chiarezza sulla sicurezza dei dispositivi interessati e sul fornire dispositivi sostitutivi ai pazienti”. Le valutazioni del rischio da parte di terzi per i dispositivi per la terapia del sonno presentate sono positive e rassicuranti e stiamo facendo notevoli progressi con il ripristino dei dispositivi interessati. “Le autorità competenti a livello globale, FDA compresa, stanno ancora esaminando i risultati dei test e le valutazioni. Condividiamo l’obiettivo comune di garantire la sicurezza del paziente e la qualità dell’assistenza sanitaria e ci impegniamo a lavorare a stretto contatto con questi organismi. Il completamento dei test e la sostituzione dei dispositivi interessati continueranno ad essere le nostre maggiori priorità”.
Risultati dei test e delle analisi per dispositivi per la terapia del sonno non esposti a pulizia con ozono
I risultati dei test e delle analisi completati per i dispositivi per la terapia del sonno di prima generazione DreamStation, System One e DreamStation Go indicano che sia improbabile che la potenziale esposizione dei pazienti al particolato espanso (PM) e ai composti organici volatili (VOC) della schiuma PE-PUR all’interno di questi dispositivi provochi danni apprezzabili alla salute dei pazienti. I test condotti e le conclusioni sono stati riassunti nella seguente tabella.
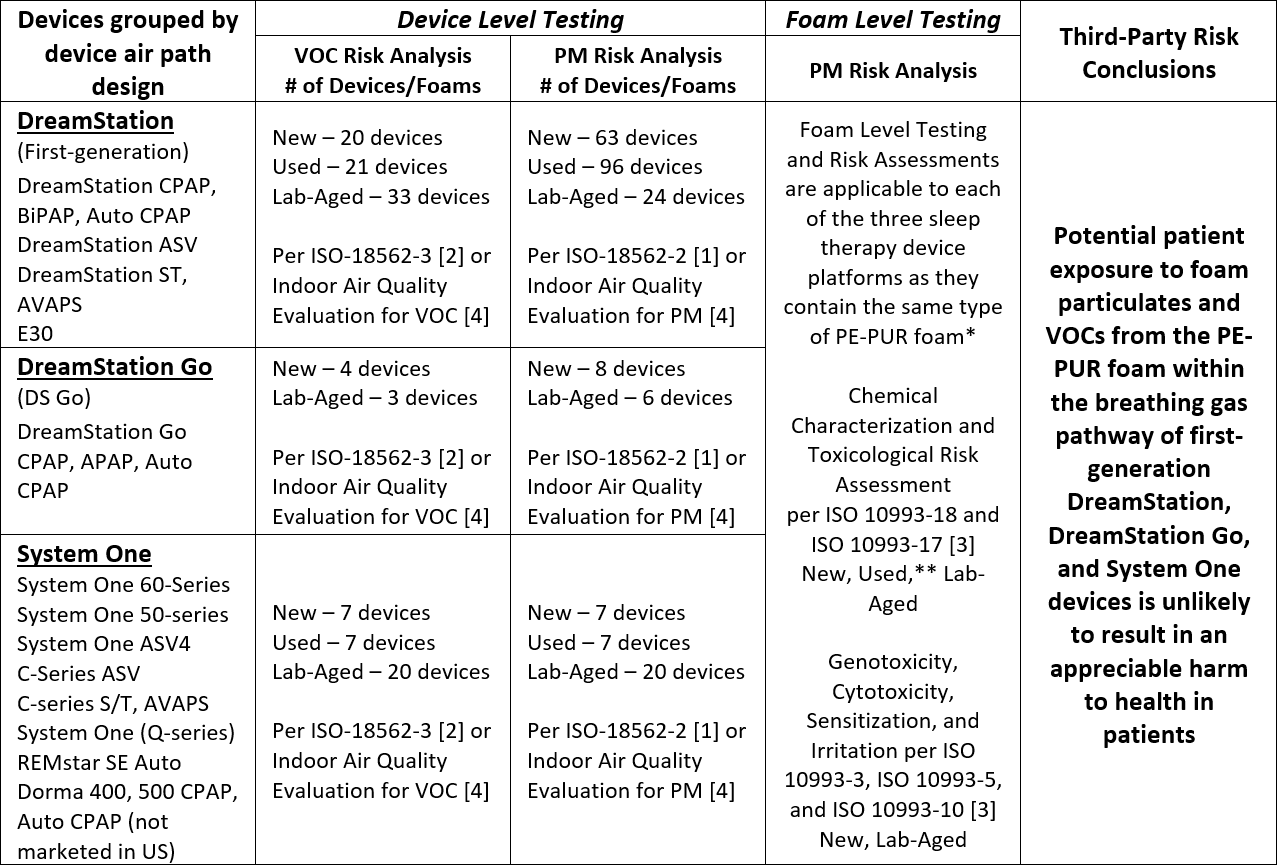
VOC: Composti Organici Volatili (VOC): PM: Particolato (PM): * La quantità totale di schiuma nei dispositivi varia da circa 1 g a 10 g, a seconda della progettazione e della configurazione del percorso d’aria del dispositivo. I dispositivi di ciascuna piattaforma condividono la stessa progettazione e la stessa configurazione del percorso di aria, compresa la quantità di schiuma presente. ** La schiuma di 7 diversi dispositivi DreamStation di prima generazione usati è caratterizzata chimicamente seguendo le norme ISO 10993-18 e -17 [3] e comprendeva schiuma rappresentativa di una serie di stati di degradazione visiva.
Test più completi e valutazioni del rischio tossicologico su diversi dispositivi con schiuma nuova, usata e invecchiata in laboratorio non hanno mostrato alcun danno apprezzabile per la salute per i VOC rilevati sulla base dei test ISO 18562-3 [2] e una valutazione del rischio da parte di terzi ha concluso che difficilmente l’esposizione ai VOC per questi dispositivi può provocare un danno apprezzabile per la salute dei pazienti.
Sono stati testati dispositivi nuovi, invecchiati in laboratorio e usati: tutti sono risultati conformi ai limiti consentiti dalla norma ISO 18562-2 [1] per le emissioni di PM. I test sono stati eseguiti su dispositivi usati (ad esempio dispositivi utilizzati in precedenza dai pazienti) con diversi gradi di degradazione (da nessuna degradazione visibile a una degradazione grave visibile), dispositivi nuovi, e dispositivi con schiuma invecchiata in laboratorio, esposti intenzionalmente a temperature (≥80 °C) e umidità (≥75% RH) significativamente elevate al fine di accelerarne la degradazione.
Inoltre, le emissioni di PM dei dispositivi usati con una visibile degradazione non sono risultate statisticamente diverse da quelle dei dispositivi usati senza degradazione, il che suggerisce che la degradazione non abbia contribuito a livelli elevati e apprezzabili di particelle respirabili all’interno dei dispositivi testati.
Anche nell’ipotesi fortemente conservativa e teorica del fatto che tutta la schiuma possa degradarsi e che il paziente venga così esposto a tutta la schiuma PE-PUR degradata all’interno del dispositivo, la valutazione del rischio effettuata da terzi ha concluso che sia improbabile che l’esposizione al particolato proveniente dalla schiuma degradata di questi dispositivi, compreso il potenziale particolato respirabile e non respirabile, comporti un danno apprezzabile per la salute dei pazienti.
Sulla base dell’ispezione visiva della schiuma nei dispositivi DreamStation di prima generazione restituiti, la prevalenza della degradazione visibile della schiuma è risultata esigua. L’ispezione visiva può solo identificare la degradazione visibile della schiuma e non è in grado di misurare la generazione di composti organici volatili o di quantificare la perdita di particolato; pertanto, sono stati eseguiti ulteriori test e analisi come descritto sopra e all’interno dell’aggiornamento completo.
Impatto della pulizia con ozono sulla degradazione della schiuma nei dispositivi DreamStation di prima generazione
Philips Respironics ha completato i test sui dispositivi DreamStation di prima generazione esposti a pulizia con ozono:
I test ISO 18562-3 VOC hanno mostrato che dopo 200 cicli di pulizia con ozono - ogni ciclo simula una notte di utilizzo e poi la pulizia con ozono - il glicole dietilenico (DEG) è diventato rilevabile come VOC. I test sono stati condotti per un massimo di 500 cicli di pulizia con ozono e il rischio tossicologico VOC di questa degradazione indotta dall’ozono ha determinato che l’esposizione alle emissioni VOC, dei dispositivi DreamStation di prima generazione sottoposti a pulizia con ozono, non comporti rischi apprezzabili per la salute dei pazienti.
Per quanto riguarda i rischi associati al particolato respirabile e non respirabile sono stati finora eseguiti dei testi su dispositivi con esposizione nota all’ozono. Ad esempio, due dispositivi DreamStation di prima generazione usati ed esposti all’ozono, secondo quanto dichiarato dall’utente, sono stati inclusi all’interno dei test sulle sostanze estraibili e rilasciabili, su cui è stata basata la valutazione del rischio tossicologico del particolato della schiuma PE-PUR del dispositivo per la terapia del sonno. L’analisi collettiva condotta da terzi ha concluso che sia improbabile che l’esposizione al particolato proveniente dalla schiuma degradata nei dispositivi DreamStation di prima generazione comporti un danno apprezzabile per la salute dei pazienti.
Come precedentemente pubblicato, i dati relativi ai dispositivi DreamStation di prima generazione, che gli utenti riferiscono essere stati puliti con ozono, rivelano che questi prodotti hanno una probabilità 14 volte maggiore di presentare una significativa degradazione visibile della schiuma/riduzione del volume rispetto ai dispositivi senza esposizione all’ozono segnalata dall’utente. Tale osservazione è in linea con i test di laboratorio, in cui i dispositivi DreamStation di prima generazione esposti a cicli crescenti di pulizia con ozono presentavano una degradazione visiva sempre più grave. Tuttavia, come già affermato, è improbabile che ciò comporti un danno apprezzabile per la salute dei pazienti.
Sintesi dei test in corso
Philips Respironics sta completando i test e le analisi rimanenti. Le valutazioni del rischio per i dispositivi System One e DreamStation Go (che contengono la stessa schiuma dei dispositivi DreamStation di prima generazione) trattati con ozono sono in fase di completamento. Per i dispositivi di ventilazione Trilogy 100/200 e OmniLab Advanced Plus, continuano i test su VOC e PM, nonché la valutazione chimica e la valutazione del rischio tossicologico. Questi dispositivi contengono un tipo di schiuma PE-PUR diverso da quello impiegato per i dispositivi DreamStation di prima generazione [5]. Philips Respironics prevede di fornire un aggiornamento in merito nel terzo trimestre del 2023.
Linee guida per operatori sanitari e pazienti
I pazienti, che attualmente utilizzano un dispositivo per la terapia del sonno non ancora riparato e non registrato, sono invitati a registrare i loro dispositivi al fine di facilitarne la riparazione.
Ai pazienti interessati che utilizzano quei dispositivi per la terapia del sonno che non sono ancora stati riparati, Philips Respironics continua a consigliare di contattare il loro medico o il loro home care provider per decidere un trattamento adeguato alla loro condizione. Tale scelta può includere l’interruzione dell’uso del dispositivo, la prosecuzione dell’uso del dispositivo interessato, l’utilizzo di un altro dispositivo simile che non faccia parte dell’avviso di sicurezza o l’utilizzo di trattamenti alternativi per l’apnea del sonno. Si consiglia inoltre ai pazienti di attenersi alle istruzioni e alle linee guida consigliate da Philips Respironics per la pulizia e la sostituzione del dispositivo per la terapia del sonno e dei relativi accessori. I prodotti per la pulizia con ozono e luce UV al momento non sono metodi di pulizia approvati per i dispositivi o le maschere per la terapia del sonno e non devono essere utilizzati.
Philips Respironics continua inoltre a raccomandare agli utenti di dispositivi di ventilazione di contattare il loro medico prima di apportare qualsiasi modifica alla terapia.
Il fondamento scientifico dei metodi di test applicati ha incluso un’attenta valutazione e mitigazione delle limitazioni dei test, ad esempio:
Note [1] ISO 18562-2: Valutazione della biocompatibilità dei percorsi dei gas respirabili in applicazioni sanitarie - Parte 2: Test per le emissioni di particolato. [2] ISO 18562-3: Valutazione della biocompatibilità dei percorsi dei gas respirabili in applicazioni sanitarie - Parte 3: Test per le emissioni di composti organici volatili [3] ISO 10993: Valutazione biologica dei dispositivi medici; Parte 1: Valutazione e test all’interno di un processo di gestione del rischio; Parte 3: Test di genotossicità, cancerogenicità e tossicità riproduttiva; Parte 5: Test di citotossicità in vitro; Parte 10: Test di irritazione e sensibilizzazione cutanea; Parte 17: Definizione dei limiti ammissibili per le sostanze rilasciabili; Parte 18: Caratterizzazione chimica dei materiali dei dispositivi medici nell’ambito di un processo di gestione del rischio. [4] La norma utilizzata per i test precedenti alla ISO 18562. [5] I dispositivi DreamStation di prima generazione, SystemOne e DreamStation Go contengono schiuma PE-PUR di tipo A, mentre i dispositivi Trilogy 100/200 contengono schiuma PE-PUR di tipo B e i dispositivi OmniLab Advanced Plus contengono schiuma PE-PUR di tipo A e B. Le differenze note tra le schiume di tipo A e di tipo B sono: la schiuma di tipo B può essere utilizzata con un adesivo acrilico sensibile alla pressione, ha una densità inferiore, ha uno spessore diverso e contiene anche un additivo per ridurre la potenziale infiammabilità.
* Notifica di ritiro volontario negli Stati Uniti/avviso di sicurezza per il resto del mondo.
Royal Philips
Philips è un'azienda leader nel settore dell’Health Technology, che ha l’obiettivo di migliorare la salute e il benessere delle persone attraverso l’innovazione. L’innovazione di Philips, incentrata sui bisogni dei pazienti e delle persone, si basa su tecnologie avanzate e profonde conoscenze cliniche e di mercato per offrire ai consumatori soluzioni per il benessere personale, e agli operatori sanitari soluzioni professionali per i loro pazienti, in ospedale e a domicilio. Con sede nei Paesi Bassi, l'azienda è leader nella diagnostica per immagini, nell’ecografia, nella terapia guidata da immagini, nel monitoraggio dei pazienti e nell'informatica sanitaria, così come nell’ambito della salute personale. Philips ha generato nel 2022 un fatturato di 17,8 miliardi di euro e impiega circa 74.000 dipendenti con vendite e servizi in oltre 100 paesi. Per maggiori informazioni: www.philips.it/newscenter
Temi
Media assets
")
")